Introduction: Unveiling the Mysteries of Apert Syndrome
Apert Syndrome, a rare genetic disorder, presents a world filled with unique challenges and complexities. Characterized by the premature fusion of skull bones, this condition impacts both the physical and mental development of those affected. But what exactly is Apert Syndrome, and how does it manifest in the lives of individuals?
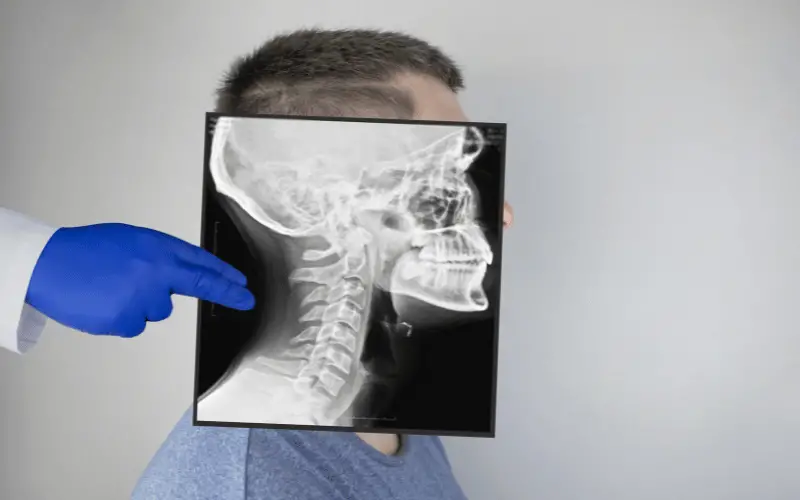
At its core, Apert Syndrome arises from genetic mutations, specifically in the FGFR2 gene. These mutations lead to the early fusion of the cranial sutures, a process known as craniosynostosis. This fusion not only shapes the distinct facial features associated with the condition but also affects brain development and function. However, the impact of Apert Syndrome extends beyond the skull. The syndrome often includes anomalies in the fingers and toes, leading to fused or webbed digits, adding to the complexity of daily activities for those affected.
The journey of understanding and managing Apert Syndrome begins with diagnosis, often detectable through prenatal testing or observed shortly after birth. Early diagnosis is crucial, paving the way for timely interventions and treatments. As we delve deeper into the world of Apert Syndrome, we uncover a spectrum of associated health complications ranging from breathing difficulties to hearing impairments and vision problems. These challenges necessitate a multidisciplinary approach to care, involving surgeries, therapies, and continuous monitoring.
But Apert Syndrome is more than a medical condition; it’s a journey that touches the lives of patients and families alike. It brings to light the importance of psychological support and community networks, emphasizing the need for awareness and understanding. As we embark on this exploration of Apert Syndrome, we aim to illuminate the paths of those living with the condition, fostering a deeper comprehension and empathy towards their experiences.
1. Genetic Roots: The Mutation Behind Apert Syndrome
Apert Syndrome, a complex genetic condition, stems from a mutation in the FGFR2 gene. This gene plays a crucial role in the development of bones and tissues in the fetus. The mutation responsible for Apert Syndrome disrupts the normal development process, leading to the premature fusion of skull bones, a condition known as craniosynostosis.
The mutation’s rarity contributes to Apert Syndrome’s uniqueness. Unlike many genetic disorders, Apert Syndrome typically arises from new mutations in the gene, meaning it often occurs in families with no prior history of the condition. This spontaneous genetic change underscores the unpredictable nature of genetic inheritance and the complexity of human genetics.
Understanding the FGFR2 mutation also opens doors to targeted treatments and interventions. As researchers delve deeper into the mechanisms of this mutation, they uncover potential pathways for therapeutic approaches, offering hope for more effective management of the condition.
The FGFR2 mutation’s impact goes beyond physical symptoms. It alters the developmental trajectory of individuals, necessitating a holistic approach to care that addresses both the physical and emotional aspects of the syndrome. This understanding emphasizes the importance of genetic counseling for families affected by Apert Syndrome, guiding them through the complexities of the condition.
In essence, the FGFR2 mutation is not just a genetic anomaly; it’s a starting point for understanding the broader implications of Apert Syndrome. It highlights the intricate relationship between our genes and our health, paving the way for advancements in both diagnosis and treatment. (1)
